Tyrosine kinase pathway
Receptor Tyrosine Kinase
Receptor tyrosine kinases (RTKs) are the high-affinity cell surface receptors for many polypeptide growth factors, cytokines, and hormones. Of the 90 unique tyrosine kinase genes identified in the human genome, 58 encode receptor tyrosine kinase proteins, distributed into 20 subfamilies. The 32 nonreceptor tyrosine kinases can be placed in 10 subfamilies. . (1) Receptor tyrosine kinases have been shown not only to be key regulators of normal cellular processes but also to have a critical role in the development and progression of many types of cancer. (2). Receptor tyrosine kinases are part of the larger family of protein tyrosine kinases, encompassing the receptor tyrosine kinase proteins which contain a transmembrane domain, as well as the non receptor tyrosine kinases which do not possess transmembrane domains. (3)
The main functions of tyrosine kinase involve the regulation of multicellular aspects of the organism. Cell to cell signals concerning growth, differentiation, adhesion, motility, and death, are frequently transmitted through tyrosine kinases. In contrast, many of the serine/threonine kinase families, such as cyclin dependent kinases and MAP kinases, are conserved throughout eukaryotes and regulate processes in both unicellular and multicellular organisms. In humans, tyrosine kinases have been demonstrated to play significant roles in the development of many disease states, including diabetes and cancer (1) Regardless of whether the TRK is membrane bound or in the cytoplasm, all tyrosine kinases regulate target protein function through transfer of phosphate from ATP to the hydroxyl group of a target protein tyrosine.
Families of Tyrosine Kinase
Epidermal growth factor receptor family - The ErbB family of proteins contains four receptor tyrosine kinases, structurally related to the epidermal growth factor receptor (EGFR), its first discovered member. In humans, the family includes Her1 (EGFR, ErbB1), Her2 (Neu, ErbB2), Her3 (ErbB3), and Her4 (ErbB4).
Fibroblast growth factor receptor (FGFR) family- The fibroblast growth factor receptors are, as their name implies, receptors that bind to members of the fibroblast growth factor family of proteins. Some of these receptors are involved in pathological conditions. For example, a point mutation in FGFR3 can lead to achondroplasia.
Vascular endothelial growth factor receptor (VEGFR) family- Vascular endothelial growth factor (VEGF), originally known as vascular permeability factor (VPF), is a signal protein produced by cells that stimulates vasculogenesis and angiogenesis.
RET receptor family- The RET proto-oncogene encodes a receptor tyrosine kinase for members of the glial cell line-derived neurotrophic factor (GDNF) family of extracellular signalling molecules.
Eph receptor family- Eph receptors (Ephs, after erythropoietin-producing human hepatocellular receptors) are a group of receptors that are activated in response to binding Eph receptor-interacting proteins (Ephrins). Ephs form the largest known subfamily of receptor tyrosine kinases (RTKs). Both Eph receptors and their corresponding ephrin ligands are membrane-bound proteins that require direct cell-cell interactions for Eph receptor activation.
Discoidin domain receptor (DDR) family- The protein encoded by this gene is a RTK that is widely expressed in normal and transformed epithelial cells and is activated by various types of collagen.
Tyrosine Kinases as Targets for Anti-Cancer Agents
The molecular pathophysiology of cancer have highlighted that many tyrosine kinases are found upstream or downstream of epidemiologically relevant oncogenes or tumor suppressor, in particular the receptor tyrosine kinases. Since tyrosine kinase forms a significant share of all oncoproteins they have taken centre stage for possible targets for cancer therapy. Hence low molecular weight tyrosine phosphorylation inhibitors (tyrphostins) have been proposed to be prospective anti-proliferating agents.
Of all the tyrosine kinase inhibitors the most successful are Gleevec, Iressa and Tarceva. The novel anticancer drug Gleevec/ Glivec/ Imatinib mesylate (Novartis STI571) is a success for CML (Chronic myeloid leukemia) and c-kit positive metastatic GIST. Gleevec kills cancer cells by turning off tyrosine kinase. In order to survive, cells need signaling through proteins (signal cascade) to keep them alive. Some of the proteins in this cascade use a phosphate group as an "on" switch. This phosphate group is added by a tyrosine kinase enzyme. In healthy cells, these tyrosine kinase enzymes are turned on and off as needed. In Ph-positive CML cells, one tyrosine kinase enzyme, BCR-Abl, is stuck on the "on" position, and keeps adding phosphate groups. Imatinib blocks this BCR-Abl enzyme, and stops it from adding phosphate groups. As a result, these cells stop growing, and even die by a process of cell death (apoptosis). Because the BCR-Abl tyrosine kinase enzyme exists only in cancer cells and not in healthy cells, imatinib works as a form of targeted therapy—only cancer cells are killed through the drug's action. In this regard, imatinib was one of the first cancer therapies to show the potential for such targeted action, and is often cited as a paradigm for research in cancer therapeutics.
What is the mechanism of action for TKI's?
Multiple kinases are involved in angiogenesis, including receptor tyrosine kinases such as the vascular endothelial growth factor receptor. Three anti-angiogenic tyrosine kinase inhibitors (TKIs), sunitinib, sorafenib and pazopanib, with differential binding capacities to angiogenic kinases were recently approved for treatment of patients with advanced cancer (renal cell cancer, gastro-intestinal stromal tumors, and hepatocellular cancer). Most small-molecule kinase inhibitors discovered to date compete with ATP. Binding of ATP to a kinase is characterized by the formation of hydrogen bonds from the adenine ring to the ATP-binding cleft of the kinase. Kinase inhibitors can target (nearby) the ATP-binding site of a kinase. The ATP-binding site is common to all protein kinases, and selectivity of kinase inhibitors is engineered by the chemical structure which is not similar to the ATP structure.(See reference 5).
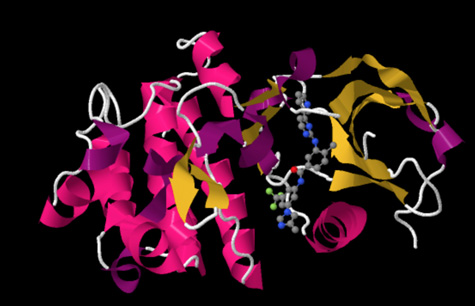
Crystal structure of the second generation Bcr-Abl tyrosine-kinase inhibitor nilotinib (red) in complex with an Abl kinase domain (blue). Nilotinib is used to treat chronic myelogenous leukemia (CML), a hematological malignancy. Structurally related to imatinib, it was developed based on the structure of the Abl-imatinib complex to address imatinib intolerance and resistance. Nilotinib is a selective Bcr-Abl kinase inhibitor that is 10-30 fold more potent than imatinib in inhibiting Bcr-Abl tyrosine kinase activity and proliferation of Bcr-Abl expressing cells. Image based on PDB 3CS9 showing 2nd Gen TKI Nilotinib in complex with Abl Kinase. Image made using Jmol.
Readings and References
1- Robinson DR, Wu YM, Lin SF (November 2000). "The protein tyrosine kinase family of the human genome". Oncogene 19 (49): 5548–57. doi:10.1038/sj.onc.1203957. PMID 11114734.
2- Zwick E, Bange J, Ullrich A (September 2001). "Receptor tyrosine kinase signalling as a target for cancer intervention strategies". Endocrine-Related Cancer 8 (3): 161–73. doi:10.1677/erc.0.0080161. PMID 11566607.
3- Hubbard SR, Till JH (2000). "Protein tyrosine kinase structure and function". Annual Review of Biochemistry 69: 373–98. doi:10.1146/annurev.biochem.69.1.373
4-Manash K. Paul and Anup K. Mukhopadhyay. "Tyrosine kinase – Role and significance in Cancer". Int J Med Sci. 2004; 1(2): 101–115.
5- Kristy J. Gotink and Henk M. W. Verheulcorresponding author. "Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action?".Angiogenesis. 2010 Mar; 13(1): 1–14.