Monoclonal Antibodies
Monoclonal antibodies (mAb or moAb) are monospecific antibodies that are identical because they are produced by one type of immune cell that are all clones of a single parent cell. Given almost any substance, it is possible to create monoclonal antibodies that specifically bind to that substance; they can then serve to detect or purify that substance. This has become an important tool in biochemistry, molecular biology and medicine. When used as medications, the non-proprietary drug name ends in -mab (see "Nomenclature of monoclonal antibodies").
Discovery
The idea of a "magic bullet" was first proposed by Paul Ehrlich who at the beginning of the 20th century postulated that if a compound could be made that selectively targeted a disease-causing organism, then a toxin for that organism could be delivered along with the agent of selectivity.
In the 1970s the B-cell cancer multiple myeloma was known, and it was understood that these cancerous B-cells all produce a single type of antibody (a paraprotein). This was used to study the structure of antibodies, but it was not yet possible to produce identical antibodies specific to a given antigen.
A process of producing monoclonal antibodies involving human-mouse hybrid cells was described by Jerrold Schwaber in 1973[1] and remains widely cited among those using human-derived hybridomas,[2] but claims to priority have been controversial. A science history paper on the subject gave some credit to Schwaber for inventing a technique that was widely cited, but stopped short of suggesting that he had been cheated.[3] The invention is generally accredited to Georges Köhler, César Milstein, and Niels Kaj Jerne in 1975;[4] who shared the Nobel Prize in Physiology or Medicine in 1984 for the discovery. The key idea was to use a line of myeloma cells that had lost their ability to secrete antibodies, come up with a technique to fuse these cells with healthy antibody-producing B-cells, and be able to select for the successfully fused cells.
In 1988 Greg Winter and his team pioneered the techniques to humanize monoclonal antibodies,[5] removing the reactions that many monoclonal antibodies caused in some patients.
Production
Hybridoma cell production
Monoclonal antibodies are typically made by fusing myeloma cells with the spleen cells from a mouse that has been immunized with the desired antigen. However, recent advances have allowed the use of rabbit B-cells. Polyethylene glycol is used to fuse adjacent plasma membranes, but the success rate is low so a selective medium is used in which only fused cells can grow. This is because myeloma cells have lost the ability to synthesize hypoxanthine-guanine-phosphoribosyl transferase (HGPRT), an enzyme necessary for the salvage synthesis of nucleic acids. This enzyme enables cells to synthesize purines by the salvage pathway, here using an extracellular source of hypoxanthine as a precursor. Ordinarily, the absence of HGPRT is not a problem for the cell because cells have an already existing biochemical pathway, the de novo pathway that they can use to synthesize purines. However, when cells are exposed to Aminopterin (a folic acid analogue, which inhibits Dihydrofolate reductase, DHFR), they are unable to use the de novo pathway and are now fully dependent on the salvage pathway for survival.
The selective culture medium is called HAT medium because it contains Hypoxanthine, Aminopterin, and Thymidine. This medium is selective for fused (hybridoma) cells. Unfused myeloma cells cannot grow because they lack HGPRT, and thus cannot replicate their DNA. Unfused spleen cells cannot grow indefinitely because of their limited life span. Only fused hybrid cells, referred to as hybridomas, are able to grow indefinitely in the media because the spleen cell partner supplies HGPRT and the myeloma partner has traits that make it immortal (as it is a cancer cell).
This mixture of cells is then diluted and clones are grown from single parent cells on microtitre wells. The antibodies secreted by the different clones are then assayed for their ability to bind to the antigen (with a test such as ELISA or Antigen Microarray Assay) or immuno-dot blot. The most productive and stable clone is then selected for future use.
The hybridomas can be grown indefinitely in a suitable cell culture media, or they can be injected in mice (in the peritoneal cavity, the gut), they produce tumors containing an antibody-rich fluid called ascites fluid. The medium must be enriched during selection to further favour hybridoma growth. This can be achieved by the use of a layer of feeder fibrocyte cells or supplement medium such as briclone. Production in cell culture is usually preferred as the ascites technique is painful to the animal and if replacement techniques exist, this method is considered unethical.
Purification of monoclonal antibodies
After obtaining either a media sample of cultured hybridomas or a sample of ascites fluid, the desired antibodies must be extracted. The contaminants in the cell culture sample would consist primarily of media components such as growth factors, hormones, and transferrins. In contrast, the in vivo sample is likely to have host antibodies, proteases, nucleases, nucleic acids, and viruses. In both cases, other secretions by the hybridomas such as cytokines may be present. There may also be bacterial contamination and, as a result, endotoxins which are secreted by the bacteria. Depending on the complexity of the media required in cell culture, and thus the contaminants in question, one method (in vivo or in vitro) may be preferable to the other.
The sample is first conditioned, or prepared for purification. Cells, cell debris, lipids, and clotted material are first removed, typically by filtration with a 0.45 µm filter. These large particles can cause a phenomenon called membrane fouling in later purification steps. Additionally, the concentration of product in the sample may not be sufficient, especially in cases where the desired antibody is one produced by a low-secreting cell line. The sample is therefore condensed by ultrafiltration or dialysis.
Most of the charged impurities are usually anions such as nucleic acids and endotoxins. These are often separated by ion exchange chromatography. Either cation exchange chromatography is used at a low enough pH that the desired antibody binds to the column while anions flow through, or anion exchange chromatography is used at a high enough pH that the desired antibody flows through the column while anions bind to it. Various proteins can also be separated out along with the anions based on their isoelectric point (pI). For example, albumin has a pI of 4.8, which is significantly lower than that of most monoclonal antibodies, which have a pI of 6.1. In other words, at a given pH, the average charge of albumin molecules is likely to be more negative. Transferrin, on the other hand, has a pI of 5.9, so it cannot easily be separated out by this method. A difference in pI of at least 1 is necessary for a good separation.
Transferrin can instead be removed by size exclusion chromatography. The advantage of this purification method is that it is one of the more reliable chromatography techniques. Since we are dealing with proteins, properties such as charge and affinity are not consistent and vary with pH as molecules are protonated and deprotonated, while size stays relatively constant. Nonetheless, it has drawbacks such as low resolution, low capacity and low elution times.
A much quicker, single-step method of separation is Protein A/G affinity chromatography. The antibody selectively binds to Protein A/G, so a high level of purity (generally >80%) is obtained. However, this method may be problematic for antibodies that are easily damaged, as harsh conditions are generally used. A low pH can break the bonds to remove the antibody from the column. In addition to possibly affecting the product, low pH can cause Protein A/G itself to leak off the column and appear in the eluted sample. Gentle elution buffer systems that employ high salt concentrations are also available to avoid exposing sensitive antibodies to low pH. Cost is also an important consideration with this method because immobilized Protein A/G is a more expensive resin.
To achieve maximum purity in a single step, affinity purification can be performed, using the antigen to provide exquisite specificity for the antibody. In this method, the antigen used to generate the antibody is covalently attached to an agarose support. If the antigen is a peptide, it is commonly synthesized with a terminal cysteine which allows selective attachment to a carrier protein, such as KLH during development and to the support for purification. The antibody-containing media is then incubated with the immobilized antigen, either in batch or as the antibody is passed through a column, where it selectively binds and can be retained while impurities are washed away. An elution with a low pH buffer or a more gentle, high salt elution buffer is then used to recover purified antibody from the support.
To further select for antibodies, the antibodies can be precipitated out using sodium sulfate or ammonium sulfate. Antibodies precipitate at low concentrations of the salt, while most other proteins precipitate at higher concentrations. The appropriate level of salt is added in order to achieve the best separation. Excess salt must then be removed by a desalting method such as dialysis.
The final purity can be analyzed using a chromatogram. Any impurities will produce peaks, and the volume under the peak indicates the amount of the impurity. Alternatively, gel electrophoresis and capillary electrophoresis can be carried out. Impurities will produce bands of varying intensity, depending on how much of the impurity is present.
Recombinant
The production of recombinant monoclonal antibodies involves technologies, referred to as repertoire cloning or phage display/yeast display. Recombinant antibody engineering involves the use of viruses or yeast to create antibodies, rather than mice. These techniques rely on rapid cloning of immunoglobulin gene segments to create libraries of antibodies with slightly different amino acid sequences from which antibodies with desired specificities can be selected.[6] These techniques can be used to enhance the specificity with which antibodies recognize antigens, their stability in various environmental conditions, their therapeutic efficacy, and their detectability in diagnostic applications.[7] Fermentation chambers have been used to produce these antibodies on a large scale.
Applications
Diagnostic tests
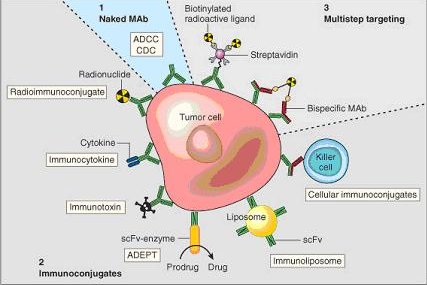
Once monoclonal antibodies for a given substance have been produced, they can be used to detect the presence of this substance. The Western blot test and immuno dot blot tests detect the protein on a membrane. They are also very useful in immunohistochemistry which detect antigen in fixed tissue sections and immunofluorescence test which detect the substance in a frozen tissue section or in live cells.Monoclonal antibodies for cancer treatment. One possible treatment for cancer involves monoclonal antibodies that bind only to cancer cell-specific antigens and induce an immunological response against the target cancer cell. Such mAb could also be modified for delivery of a toxin, radioisotope, cytokine or other active conjugate; it is also possible to design bispecific antibodies that can bind with their Fab regions both to target antigen and to a conjugate or effector cell. In fact, every intact antibody can bind to cell receptors or other proteins with its Fc region.
The illustration below shows all these possibilities:
MAbs approved by the FDA include[9]
Chimeric and humanized antibodies
One problem in medical applications is that the standard procedure of producing monoclonal antibodies yields mouse antibodies. Although murine antibodies are very similar to human ones there are differences. The human immune system hence recognizes mouse antibodies as foreign, rapidly removing them from circulation and causing systemic inflammatory effects. Such responses are recognised as producing HACA (Human Anti-Chimeric) antibody antibodies or HAMA (Human Anti-Mouse) antibodies.
A solution to this problem would be to generate human antibodies directly from humans. However, this is not easy, primarily because it is generally not seen as ethical to challenge humans with antigen in order to produce antibody; the ethics of doing the same to non-humans is a matter of debate. Furthermore, it is not easy to generate human antibodies against human tissues.
Various approaches using recombinant DNA technology to overcome this problem have been tried since the late 1980s. In one approach, one takes the DNA that encodes the binding portion of monoclonal mouse antibodies and merges it with human antibody-producing DNA. One then uses mammalian cell cultures to express this DNA and produce these half-mouse and half-human antibodies. (Bacteria cannot be used for this purpose, since they cannot produce this kind of glycoprotein.) Depending on how big a part of the mouse antibody is used, one talks about chimeric antibodies or humanized antibodies.
'Fully' Human Monoclonal Antibodies
Ever since the discovery that monoclonal antibodies could be generated in-vitro, scientists have targeted the creation of 'fully' human antibodies to avoid some of the side effects of humanised and chimeric antibodies. Two successful approaches were identified - phage display-generated antibodies and mice genetically engineered to produce more human-like antibodies.
One of the most successful commercial organisations behind therapeutic monoclonal antibodies was Cambridge Antibody Technology (CAT). Scientists at CAT demonstrated that phage display could be used such that variable antibody domains could be expressed on filamentous phage antibodies. This was reported in a key Nature publication[10].
Other significant publications include:
- D. Marks, H.R. Hoogenboom, T.P. Bonnert, J. McCafferty, A.D. Griffiths and G. Winter (1991) "By-passing immunisation. Human antibodies from V-gene libraries displayed on phage." J.Mol.Biol., 222, 581-597.
- S. Carmen and L. Jermutus, "Concepts in antibody phage display". Briefings in Functional Genomics and Proteomics 2002 1(2):189-203[11]
CAT developed their display technologies further into several, patented antibody discovery/functional genomics tools which were named ProximolTM[12] and ProAbTM. ProAb was announced in December 1997[13] and involved highthroughput screening of antibody libraries against diseased and non-diseased tissue, whilst Proximol used a free radical enzymatic reaction to label molecules in proximity to a given protein[14][15].
Genetically engineered mice, so called transgenic mice, can be modified to produce human antibodies[16], and this has been exploited by a number of commercial organisations:
- Medarex - who market their UltiMab platform[17]
- Abgenix - who marketed their Xenomouse technology. Abgenix were acquired in April 2006 by Amgen[18].
- Regeneron's VelocImmune technology[19].
Monoclonal antibodies have been generated and approved to treat: cancer, cardiovascular disease, inflammatory diseases, macular degeneration, transplant rejection, multiple sclerosis, and viral infection (see monoclonal antibody therapy).
In August 2006 the Pharmaceutical Research and Manufacturers of America reported that U.S. companies had 160 different monoclonal antibodies in clinical trials or awaiting approval by the Food and Drug Administration.[20]
Examples
| Type | Application | Mechanism | Mode |
|---|---|---|---|
| infliximab |
|
inhibits TNF-α | chimeric |
| basiliximab |
|
inhibits IL-2 on activated T cells | chimeric |
| bevacizumab |
|
inhibits VEGF | humanized |
| abciximab |
|
inhibits the receptor GpIIb/IIIa on platelets | chimeric |
| daclizumab |
|
inhibits IL-2 on activated T cells | humanized |
| gemtuzumab |
|
targets an antigen on leukemia cells | humanized |
| alemtuzumab |
|
targets an antigen CD52 on T- and B-lymphocytes | humanized |
| rituximab |
|
targets phosphoprotein CD20 on B lymphocytes | chimeric |
| palivizumab |
|
inhibits an RSV protein | humanized |
| trastuzumab |
|
targets the HER2/neu (erbB2) receptor | humanized |
| etanercept |
|
contains TNF receptor | fusion protein |
| adalimumab |
|
inhibits TNF-α | human
|
| Nimotuzumab |
|
EGFR inhibitor | Humanized |
References
- Schwaber, J and Cohen, E. P., "Human x Mouse Somatic Cell Hybrid Clones Secreting Immunoglobulins of Both Parental Types," Nature, 244 (1973), 444--447.
- Science Citation Index
- Alberto Cambrosio Peter Keating, Journal of the History of Biology. Between fact and technique: The beginnings of hybridoma technology, Volume 25,Issue 2,175- 230.
- Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975;256:495-7. Reproduced in J Immunol 2005;174:2453-5.
- Riechmann L, Clark M, Waldmann H, Winter G. Reshaping human antibodies for therapy. Nature 1988;332:323-7.
- Siegel DL (2002). "Recombinant monoclonal antibody technology". Transfusion clinique et biologique : journal de la Société française de transfusion sanguine 9 (1): 15–22.
- Schmitz U, Versmold A, Kaufmann P, Frank HG (2000). "Phage display: a molecular tool for the generation of antibodies--a review". Placenta 21 Suppl A: S106–12.
- Modified from Carter P: Improving the efficacy of antibody-based cancer therapies. Nat Rev Cancer 2001;1:118-129
- Takimoto CH, Calvo E. "Principles of Oncologic Pharmacotherapy" in Pazdur R, Wagman LD, Camphausen KA, Hoskins WJ (Eds) Cancer Management: A Multidisciplinary Approach. 11 ed. 2008.
- http://www.nature.com/nature/journal/v348/n6301/abs/348552a0.html
- http://bfgp.oxfordjournals.org/cgi/content/abstract/1/2/189
- http://www.ncbi.nlm.nih.gov/pubmed/11968489
- http://www.ukbusinesspark.co.uk/cay92125.htm
- http://www.sciencedirect.com/science?
- http://www.chidb.com/newsarticles/issue3_1.ASP
- http://www.ncbi.nlm.nih.gov/pubmed/7494109
- http://www.medarex.com/Development/UltiMAb.htm
- http://wwwext.amgen.com/media/media_pr_detail.jsp?year=2006&releaseID=837754
- http://www.regeneron.com/velocimmune.html
- PhRMA Reports Identifies More than 400 Biotech Drugs in Development. Pharmaceutical Technology, August 24, 2006. Retrieved 2006-09-04.
- Rang, H. P. (2003). Pharmacology. Edinburgh: Churchill Livingstone. pp. 241, for the examples infliximab, basiliximab, abciximab, daclizumab, palivusamab, palivusamab, gemtuzumab, alemtuzumab, etanercept and rituximab, and mechanism and mode.
- Olson, Wayne P. Separations Technology: Pharmaceutical and Biotechnology Applications. Buffalo Grove, IL: Interpharm Press, Inc., 1995.
- Shepherd, Philip. Christopher Dean. Monoclonal Antibodies. New York: Oxford University Press, 2000.